
In the rapidly evolving pharmaceutical industry, regulatory submissions play a critical role in the drug approval process. Companies must ensure that their documentation meets the stringent requirements set by regulatory bodies worldwide. The Electronic Common Technical Document (eCTD) has emerged as the gold standard for submitting applications for drug approvals, facilitating a more structured and efficient approach to compliance. However, navigating the complexities of eCTD requirements can be daunting, especially for companies operating across multiple regions.
A Real-World Scenario
Consider a mid-sized pharmaceutical company developing an innovative treatment for a rare disease. While their research and clinical trials were promising, their regulatory submission process faced significant hurdles. The team struggled with manual document management, inconsistent formatting, and repetitive submission rejections due to non-compliance with agency guidelines. To overcome these challenges, they adopted an advanced eCTD submission tool, E-CTD Plus. The transition not only streamlined their regulatory submissions but also improved eCTD validation, reducing errors and accelerating their drug approval process.
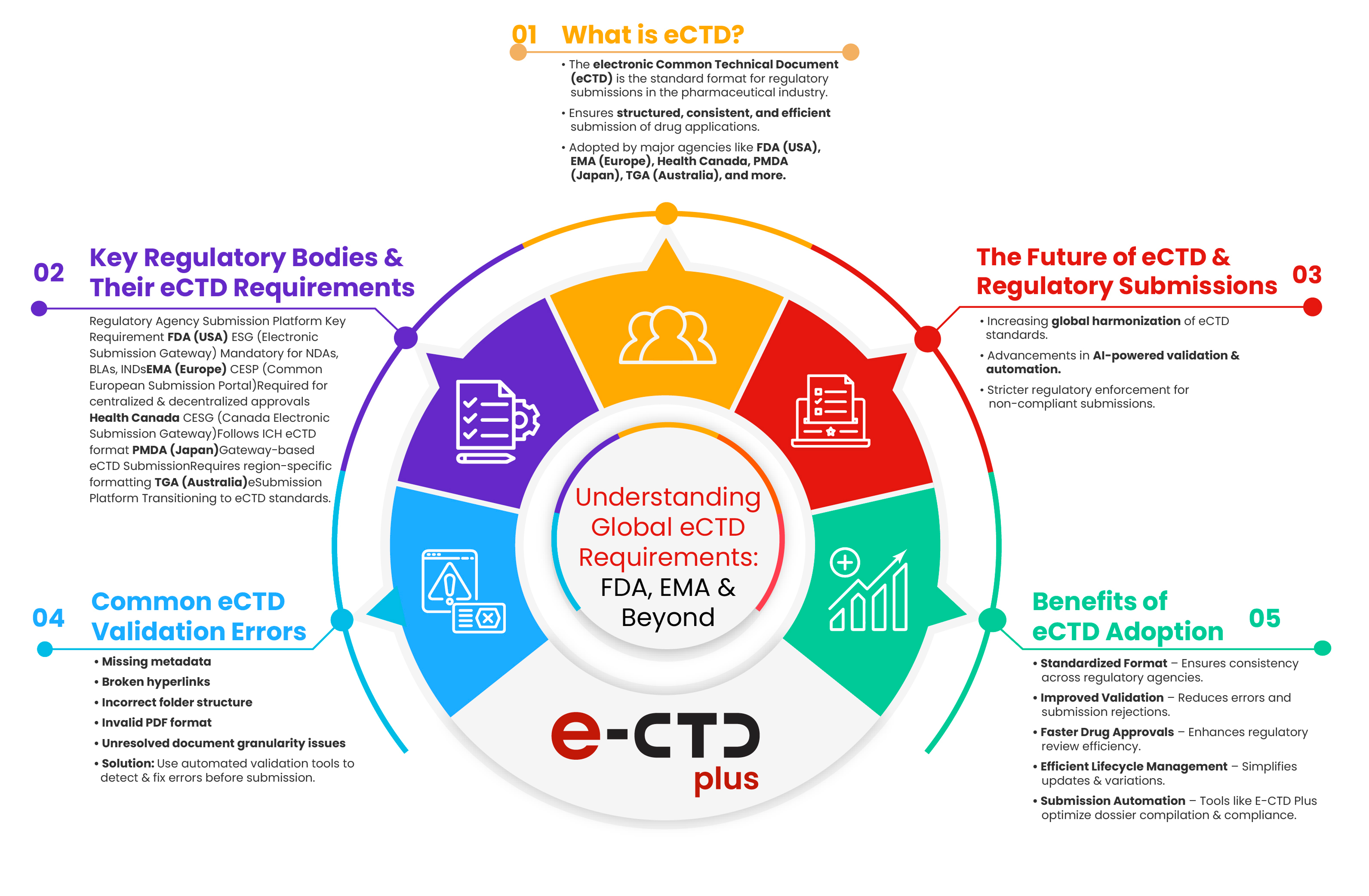
Overview of eCTD Requirements by Major Regulatory Bodies
The eCTD format is widely accepted across global regulatory agencies, but specific requirements vary. Below is an overview of the key differences and similarities between the FDA and EMA requirements:
U.S. Food and Drug Administration (FDA)
- Submission Format: Mandatory for New Drug Applications (NDAs), Biologics License Applications (BLAs), and Investigational New Drug (IND) applications.
- Validation Criteria: Requires adherence to structured metadata, document granularity, and regional requirements outlined in the eCTD Module 1 specifications.
- Lifecycle Management: Emphasizes compliance with sequence numbering, submission tracking, and version control.
European Medicines Agency (EMA)
- Submission Format: Mandatory for all centralized, decentralized, and mutual recognition procedures.
- Validation Standards: Requires submission via the Common European Submission Platform (CESP) or the EMA Gateway.
- Lifecycle Management: Focuses on dossier lifecycle tracking, electronic signatures, and interoperability with national agencies.
Beyond FDA and EMA: Other Regulatory Agencies
- Health Canada: It implements eCTD requirements similar to the FDA but with unique validation rules.
- Japan’s PMDA: Requires region-specific formatting and validation tools.
- TGA (Australia): Transitioning to eCTD to align with international standards.
The Importance of eCTD Validation
Ensuring compliance with regulatory guidelines hinges on robust eCTD validation. Common issues, such as missing hyperlinks, incorrect metadata, and structural inconsistencies can result in submission delays. Advanced validation tools help detect these errors before submission, improving regulatory approval timelines.
The Role of Submission Automation in Enhancing Efficiency
Submission automation has revolutionized how pharmaceutical companies handle regulatory submissions. Tools like E-CTD Plus integrate with existing document management systems, automating key tasks such as:
- Metadata Management: Reducing manual input errors.
- Real-Time Validation: Identifying issues before submission.
- Dossier Compilation: Ensuring structured and compliant CTD dossier submissions.
By leveraging submission automation, companies can accelerate their drug-approval process while maintaining pharmaceutical compliance.
Frequently Asked Questions (FAQs)
The eCTD format ensures structured documentation, improves review efficiency, and enhances compliance with regulatory requirements.
Proper validation prevents submission rejections, reduces delays, and ensures faster regulatory assessments.
While both agencies mandate eCTD submissions, the FDA emphasizes sequence tracking, while the EMA focuses on centralized digital submission via CESP.
Yes, automated tools help manage metadata, validate submissions, and streamline the compilation of CTD dossiers.
E-CTD Plus simplifies dossier preparation, enhances validation, and automates submission tracking, ensuring regulatory compliance.
Yes, regulatory agencies continually update their guidelines to enhance digital submission efficiency and harmonization across regions.
Conclusion
By understanding global eCTD requirements and leveraging submission automation tools, pharmaceutical companies can navigate regulatory complexities more efficiently, ensuring faster drug approvals and sustained compliance.